
Understanding what causes colorectal cancer (CRC) could help to combat this disease of the colon. Writing in Nature, Pleguezuelos-Manzano et al.1 report evidence that strengthens a previously suspected connection to a type of gut bacterium. The authors implicate this microbe by pinpointing bacterial ‘fingerprints’ in DNA alterations found in CRC cells.
Certain bacteria produce genotoxic molecules — those capable of damaging DNA. These molecules can cause mutations if, for example, mistakes occur during the DNA-repair process that tries to fix genotoxic damage. In 2006, a genotoxin called colibactin, which is made by certain strains of the gut-dwelling bacterium Escherichia coli, was discovered2. That original description also shed light on how colibactin is produced by E. coli, identifying a key region of bacterial DNA, termed the pks island (the microbes that have this island are called pks+ E. coli), which encodes various components of an ‘assembly line’ that makes colibactin.
By producing colibactin, pks+ E. coli can accelerate tumour formation in animal models3. Moreover, these bacterial strains are more prevalent in close association with the epithelial cells in the mucosa of the colon in people who have CRC than in those who don’t3. However, the complexity of the colibactin-producing assembly line and the molecule’s considerable instability pose substantial challenges to researchers trying to decode the workings of the pks island and to characterize colibactin’s structure.
There are several questions to be answered. For example, what is the mechanism of action of colibactin? What types of change might it make to DNA nucleotides? And does colibactin activity have relevance to human cancer? It is known4 that pks+ E. coli damages the DNA of cells it infects by causing adenine nucleotides to undergo a type of modification called alkylation. Subsequent evidence proposing a symmetrical colibactin structure indicates that the molecule has two ‘warheads’ made of a structure called cyclopropane, which target adenines5. How common pks+ E. coli is in the gut of human populations is not fully known.
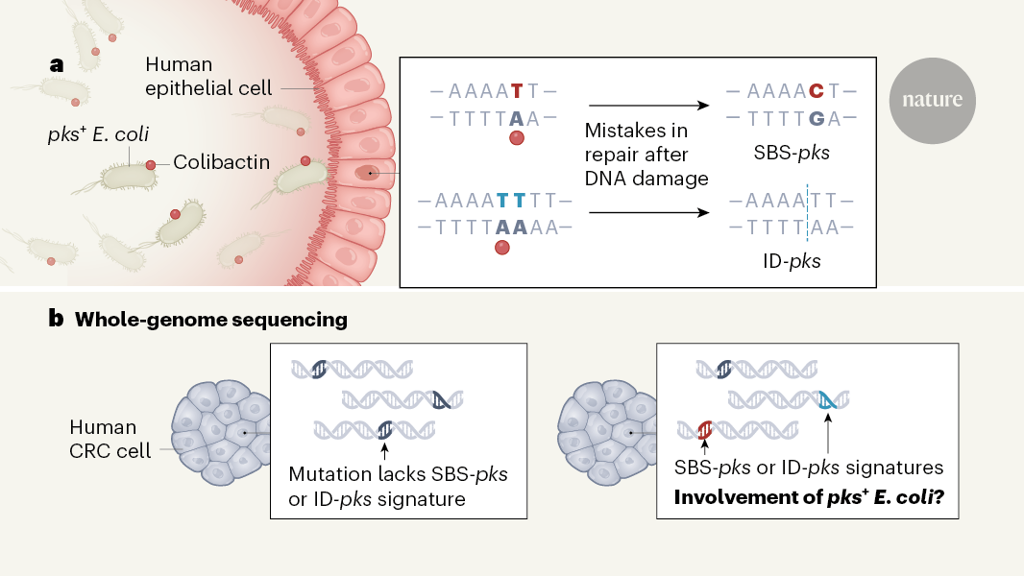
To determine the details of DNA changes that might be induced by pks+ E. coli, Pleguezuelos-Manzano and colleagues turned to a ‘mini-gut’ cellular system that mimics the human intestine (Fig. 1). This approach uses a clump of human epithelial cells grown in vitro called an organoid or, specifically, a colonoid, because it is made of colon cells. The authors exposed colonoids either to pks+ E. coli isolated from a person with CRC or to an engineered version of the bacterium that did not make colibactin. This set-up enabled the bacteria to interact with the type of cellular surface they would encounter in the lumen of the colon. Whole-genome sequencing of colonoid cells enabled the authors to compare the mutations in cells exposed to E. coli that produced colibactin or that were defective in producing it.

Figure 1 | Specific DNA mutations that can be induced by a bacterium also occur in human cancer. Pleguezuelos-Manzano et al.1 report the discovery of types of DNA mutation caused by a gut-dwelling bacterium. a, The authors used a model of the human gut consisting of epithelial cells grown in vitro and exposed these cells to Escherichia coli bacteria that make the molecule colibactin (such microbes are termed pks+ E. coli). Colibactin modifies DNA at adenine (A) nucleotides. This results in DNA damage, and errors in the repair process lead to mutations. Pleguezuelos-Manzano et al. identified specific types of mutation induced by colibactin. One type was single-base substitutions (called SBS-pks), typically identified as the replacement of a thymine nucleotide with a different nucleotide, leading to a different base pair at that site. Another type was insertion or deletion mutations (ID-pks), characterized by the deletion of thymine (T) nucleotides in a string of thymines. b, Using whole-genome sequencing data already available for cells from human colorectal cancer (CRC), the authors analysed the mutations present, and identified a subset of tumours that had mutations characteristic of colibactin exposure. This raises the question of whether these mutations arose because of exposure to pks+ E. coli.
From this analysis, the authors determined a unique colibactin mutational signature — specific patterns of DNA alterations that arose in the presence of colibactin. This signature predominantly included two types of change. One type is the substitution of a single DNA nucleotide base for a different nucleotide (single-base substitutions, termed SBS-pks). These were skewed towards a change described as T→N, in which a thymine (T) nucleotide is replaced by any other type of nucleotide (N).
The other type of change is a small insertion or deletion of nucleotides, characterized by deletions of single nucleotides in stretches of thymine nucleotides (known as T homopolymers). This sort of alteration is termed ID-pks. Interestingly, both SBS-pks and ID-pks occur preferentially downstream of adenine nucleotides, consistent with the proposed mode of action of colibactin, with two warheads targeting adenine nucleotides that are located in close proximity on opposite strands of the DNA (one warhead targets an adenine upstream of the site of damage and the other targets the site of damage)4,5.
To determine whether this colibactin-associated mutational signature might be relevant to human disease, the researchers analysed a data set6 of whole-genome sequences for 496 human CRC tumours that had migrated from their primary site in the colon to form secondary growths termed metastases. Remarkably, the authors found that SBS-pks and ID-pks mutations were present in 7.5% and 8.8%, respectively, of CRC metastases, which is more frequent than in metastases of cancers of other primary origins. For example, SBS-pks and ID-pks mutations were found in 2.1% and 4.2%, respectively, of metastases of urinary-tract cancers, and in 1.6% and 1.6%, respectively, of head and neck tumour metastases. This pattern is consistent with the probability of exposure to pks+ E. coli at these different body sites, considering that the urinary tract, head and neck are only occasionally exposed to E. coli. When the authors assessed 2,208 predominantly primary CRC tumours from an independent data set (see go.nature.com/3d6utsx), 5.0% and 4.4% of the tumours, respectively, had high SBS-pks and ID-pks signatures, which supports the idea that pks+ E. coli are involved in the early stages of tumour formation.
Further incriminating evidence is the observation that 2.4% of the most common CRC ‘driver mutations’ (those implicated in having a causal role in the disease), and 5.3% of mutations in the gene APC, which is frequently mutated in CRC, match the SBS-pks or ID-pks target motifs. Interestingly, SBS-pks- and ID-pks-like mutational signatures that are thought to arise from a mutational event in early childhood are found in the ‘crypt’ region of healthy human colons7, suggesting that exposure to pks+ E. coli early in life might be a causative event that contributes to cancer formation. Together, these findings depict a potential pathway by which a genotoxic bacterium could contribute to the development of cancer.
Pleguezuelos-Manzano and colleagues’ work fills an important gap in our knowledge regarding the mutational consequence of exposure to pks+ E. coli in the context of cancer formation. However, some issues should be considered before sounding the alarm bells on pks+ E. coli, conducting screening tests to identify people who have these bacteria or initiating procedures to eradicate the microbes.
Of note, one widely used probiotic strain of E. coli (a strain used with the aim of promoting health) is the Nissle 1917 strain that carries the pks island and requires the strain’s DNA-damaging activity for its inflammation-attenuating activity in animal models8. This probiotic strain has a long history of safety in its use for human treatment9, which suggests that the cancer-inducing activity of pks+ E. coli is context-dependent. Although the mini-gut system is a powerful tool for investigating interactions between bacteria and human cells, it bypasses numerous other layers of interaction in the gut, such as the mucus barrier, cells of the gut immune system, and other types of microbe. Any of these components might influence the ability of pks+ E. coli to attach to epithelial cells — an absolute requirement for colibactin to have a genotoxic effect2.
Indeed, mice colonized with pks+ E. coli rarely develop colon tumours in the absence of factors such as inflammation3,10, cancer-initiating mutations3,10,11 or other cancer-promoting bacteria11. The fact that pks+ E. coli are found in approximately 67% of people who have CRC3, yet the mutational signature was observed in only about 5% of the primary CRC tumours as shown by Pleguezuelos-Manzano et al., supports the concept of context-dependent genotoxicity. It will be crucial to define the interplays between human and bacterial cells and between different bacterial cells that might lead to conditions that would permit cancer formation mediated by pks+ E. coli. In addition, determining the evolution of this mutational signature in relation to tumour development over time in animal models would strengthen the evidence supporting a direct cause-and-effect relationship.
This work also raises the question of what mutational signatures might be induced by other microbial genotoxins. For example, a toxin called cytolethal-distending toxin, which is produced by certain Campylobacter bacteria, damages DNA and promotes colon-tumour development in mice12. Comparing the mutational signatures arising from colibactin and other microbial genotoxins13 might offer a clearer picture of the mechanisms that can drive cancer formation and the potential synergistic effects that might arise. Moreover, obtaining a comprehensive catalogue of the distinctive DNA ‘scars’ left by encounters with various microorganisms might be useful for tracking down possible culprits that drive tumour formation.
Pleguezuelos-Manzano et al. have highlighted the potential contribution of colibactin to cancer formation in humans, thereby adding further evidence that the community of gut microorganisms contains members with possible cancer-promoting properties. There is growing interest in manipulating gut bacteria for therapeutic purposes, and one could consider investigating strategies against pks+ E. coli strains in humans once the causal link to disease is firmly established.